 Testimonials
Testimonials
Annex 1 Revision 2020
+ Book Online Now
Expanding from 127 clauses in the 2008 revision to 300 clauses in the 2020 revision, will Annex 1 be any less ambiguous? Clearly the latest consultation draft is more than a revision, it is a rewrite.
The revised guidance requires companies to formally document a contamination control strategy which considers all the technical and organisational measures in place to ensure sterile products meet their critical quality attributes. This review document is intended to provide a summary of major changes and enhanced requirements in an easy to read format.
Keywords: Quality Risk Management (QRM), Contamination Control Strategy (CSS), Pharmaceutical Quality System (PQS) and Risk Assessment, Barrier Technologies, Clean Air and Clean Air Equipment Qualification and Classification, Transfer of Materials and Personnel, Disinfection (was Sanitation), Equipment, Utilities (4 from 2), Personnel, Production and Specific Technologies, Aseptic Processing and Preparation, Sterilisation, Filtration, Lyophilisation, Closed Systems, Single Use Systems, Viable and Non-Viable Environmental and Process Monitoring, Viable Particle Monitoring (Microbial Monitoring), Aseptic Process Simulation (APS or Media Fill), Quality Control (QC).
Author – Marcus Booth, Honeyman Training ©2020
Introduction

Within the pharmaceutical industry, the manufacture of sterile medicinal products are considered by most to be high risk operations due to the risk of product contamination and subsequent risk to the patient from administration with a product which is not fit for its intended use. The risk of a non-sterile product reaching the patient is a real risk which has resulted the loss of patient lives during several unwanted events including the Davenport incident in 1971, and the New England Compounding Centre in 2012. This has resulted in the continued need for regulation around the manufacture of sterile medicinal products and the need for a dedicated regulatory guideline to define the minimum requirements for pharmaceutical manufacturers to follow in the form of Annex 1 to the main GMP guide.
Currently Annex 1 is under revision with the first consultation draft having been issued in 2017 for public review and comment. The revision committee responsible for the latest draft comprises the EMA, WHO and PICS and includes FDA input to produce a harmonised guideline. Since the first draft was issued industry provided a significant number of comments which have resulted in a second draft being issued in February 2020 to incorporate the comments from the original draft. There now follows a further period of public consultation and a final committee review before issue of the final document. During the last 12 years since the 2008 version there have been considerable advances to technology and changes to the way industry operates resulting in the GMP guideline becoming outdated, hence the need for a revision. The latest consultation draft is more than a revision to the document it is a re-write with significant changes throughout the document including new sub-chapters and enhancement of existing long-standing requirements.
The latest consultation draft is a re write
The task for the regulatory committee preparing the guideline is to produce a document which sets out the minimum requirements for GMP compliance which every manufacturer who intends to sell their products into Europe must follow or justify an alternative approach. The objective is not to achieve the perfect document, but to achieve consensus on the final text before issue. The text needs to be open ended enough to accommodate the introduction of new technologies and allow manufacturers the flexibility to adopt alternative ways of working, whilst still meeting the fundamental GMP principles, therefore it cannot be overly prescriptive.
The issue with the GMP guidelines comes down to interpretation of the text and the individual clauses. The interpretation of the clauses in turn comes down to the experiences, cultural background and perspective of the individual or organisation reading it and how the changes are perceived to affect their manufacturing operations. Major changes to regulations or a lack of clarity around the interpretation of enhanced requirements could result in the need for modifications to facilities or manufacturing operations or result in deficiencies at the next GMP inspection so it becomes a very emotive subject. This document is not intended to provide detailed scrutiny of the wording adopted in the individual clauses of the latest draft of the guideline, but rather to provide a high level summary of the major changes and enhanced requirements in an easy to read format.
The two most significant changes to the document are the requirement for companies to manufacture sterile products using the principles of Quality Risk Management (QRM) and implement a Contamination Control Strategy across the facility.
The two most significant changes to the document are the requirement for companies to manufacture sterile products using the principles of Quality Risk Management (QRM) and implement a contamination control strategy across the facility. This is an example of where the current 2008 Annex 1 text has become outdated because the use of QRM principles in decision making within pharmaceutical manufacturing has been a regulatory requirement for several years. At Honeyman Training we have been training on implementing contamination control strategies for several years using risk assessment tools like HACCP in our Critical Factors for Sterile Product Manufacturing Course and we see many companies have completed these assessments, so the regulations are being updated to reflect current industry practice. The revision to Annex 1 now provides a structure for the contamination control strategy (CCS) and examples of when and where to apply risk management principles and these are being examined closely by industry during the consultation period.
Contamination Control Strategy (CCS)
In the revised Annex the principle behind a CCS is defined. The recommended approach to the CCS follows basic HACCP principles where the product and process is reviewed in its entirety to understand each unit operation and to assess the potential contamination hazards and at each step and the measures in place to prevent the hazard occurring. The hazards are clearly defined in the guideline (microbial, endotoxins and particulate matter). The original scope of Annex 1 was for sterile products only, however the scope of the document has been increased whereby the principles defined in the document could be applied to non-sterile products. So really the same HACCP principles could be applied to assess the risk of product contamination and cleaning validation in the manufacture of non-sterile products although the risk assessment tools would need to be modified for the purpose. The revised Annex outlines the elements to consider in a documented CCS strategy which include a mix of technical measures in place including the facility, equipment design and materials in conjunction with a series of organisational measures including quality systems, supplier and equipment qualification and process monitoring.
A summary of the elements of a CCS are included in Table 1. It goes without saying that the caveat here is that these are the basic elements to considered within a documented CCS, but the CCS should not be limited to just these elements. This highlights the open-ended nature of the Annex and that the intention is to provide the minimum set of requirements.
Table 1 Technical and Organisational Elements of a CCS
| Technical Measures | Organisational Measures |
| Design of the plant and process | Vendor Approval |
| Premises and Equipment | Process Risk Assessment |
| Utilities | Process Validation |
| Raw Materials Control | Preventative Maintenance |
| Product Containers and Closures | Quality SystemsReview of TrendsCAPA, Root Cause & InvestigationUse of Investigational Tools |
| Outsourced Services | |
| Cleaning & Disinfection | |
| Monitoring Systems |
Pharmaceutical Quality System (PQS) & Risk Assessment
In terms of organisational measures, the requirements for a quality system have been significantly revised in the latest revision with a dedicated section on the PQS. Historically and currently the highest number of observed inspection deficiencies are attributed to inadequacies of the PQS and documentation of events and investigations. On review of MHRA 2018 inspection deficiency data this is evident with 1,500 deficiencies relating to non- compliance with Chapter 1.
Graph 1 GMP Inspection Deficiencies by Chapter in 2018
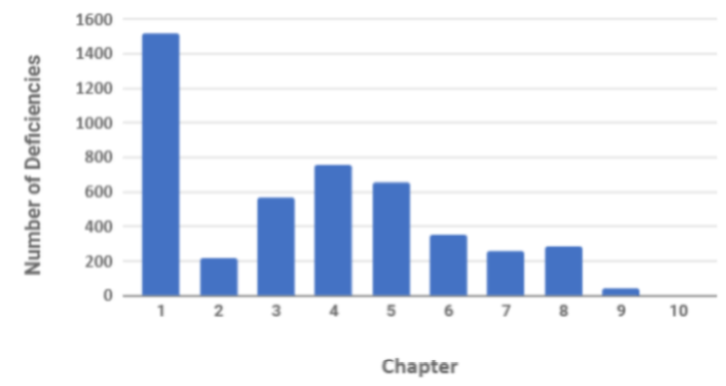
(Source Table Presentation: Unger Consulting Inc ref https://www.bioprocessonline.com/doc/an-analysis-of-mhra-s-annual-gmp-inspection-deficiencies-report-0001; Source Table Data MHRA)
In terms of deficiencies relating to technical aspects then deficiencies related to Annex 1 Sterile products, Annex 15 Validation and Annex 16 QP release were the most frequently observed.
Graph 2 GMP Inspection Deficiencies by Annex in 2018
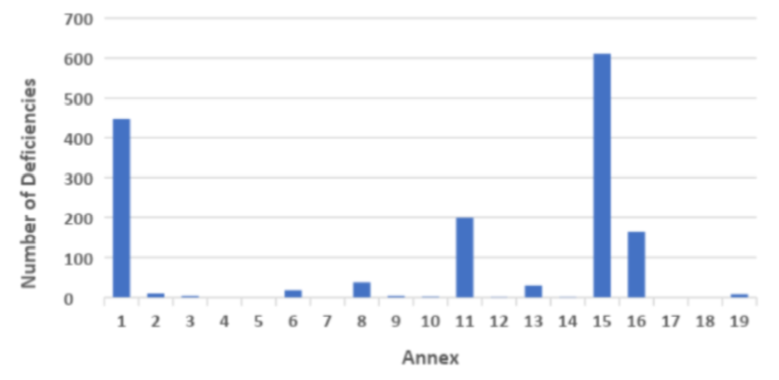
(Source:Table Presentation Unger Consulting Inc ref https://www.bioprocessonline.com/doc/an-analysis-of-mhra-s-annual-gmp-inspection-deficiencies-report-0001; Source Table Data MHRA)

The new section on the PQS clarifies the expectations for an effective quality system which includes ensuring that the organisation has sufficient knowledge and expertise in relation to the products they are manufacturing and that decisions are documented. This requirement appears to be obvious but it can be challenging for companies to retain staff due to buoyancy within the industry and of course when the staff leave the knowledge is lost unless the decision making process has been documented. The underlying message here is to understand the product and process in enough detail to be able to effectively assess the hazards to product quality from contamination and ensure control measures are in place which proactively mitigate the risk of the product becoming contaminated.
The underlying message here is to understand the product and process in enough detail to be able to effectively assess the hazards to product quality from contamination and ensure control measures are in place which proactively mitigate the risk of the product becoming contaminated.
This is achieved through a series of measures starting with effective design, qualification and validation of the facility and equipment in conjunction with an appropriate number of suitably qualified staff with right attitude and experience to be able to manage the process effectively. During manufacture of the product to have systems in place to monitor and trend the performance of the process and to have systems in place investigate process deviations which may impact product quality using a documented system of risk assessment. For the organisation to be able to act upon the data from the risk assessment to correct and prevent the deviation occurring again and for the Qualified Person to be able to make effective batch deposition decisions effectively. Once the CCS is implemented the maintenance of it will become part of the periodic product quality review to confirm that any changes to any part of the process have been implemented in accordance with the CCS and GMP. There is also greater emphasis on assessing all aspects of the product lifecycle including supply of raw materials and components as well as transportation and storage of the finished products during supply to the market.
Premises
This section is focussed on the technical aspects of cleanroom design and operation including the use of barrier technology, transfer of people / materials cleanroom, cleaning and disinfection and demonstrating that the maintenance of suitable environmental conditions during qualification of the cleanroom.
Barrier Systems

There are several significant revisions to this section and the first being that companies are required to consider the use of Barrier technology (RABS or isolators) as part of their CCS.
Few would argue that barrier technology provides a greater level of protection of the product from microbial contamination provided they are effectively managed, however there are many different barrier system configurations (Open/ Closed systems) and the document does not offer any guidance on how to select an appropriate system to protect the particular product manufactured.
For barrier technologies consideration must be given to the entry of materials / components into the critical zone whilst maintaining the Grade A environment. The CCS should include an assessment of the material transfer process and if required, the decontamination methods. Also, consideration should be given to the background environment for RABS and isolators depending on if closed or open systems are utilised. The guideline also makes the point that negative pressure isolators should only be used when containment of the product is considered essential and appropriate risk control measures are applied.
Requirements for frequency of integrity testing of the barrier system and leak testing of gloves are made (minimum of at the start and end of batch and after each intervention which may affect the integrity of the system) and the frequency of glove replacement should be defined in the CCS.
Requirements for disinfection of RABS and Isolators are made (automated methods for Isolators) and the application of a sporicidal agent for RABS (as most are open systems) and evidence must be available to demonstrate the disinfectant agent does not impact the product.
Clean Air Supply
There are several clauses related to the air supply to the cleanroom which is justified as this is one of the key control measures on achieving the required environmental conditions for manufacturing. These include ensuring the cleanroom is supplied with filtered air, maintenance of positive pressure to adjacent rooms of different grades (minimum of 10 pascals) and effective monitoring and warning systems in the event of a failure or reduction in pressure differentials between cleanrooms/ isolators. There are numerous references to the importance of airflow visualisation throughout the document to verify that the air flow within the cleanroom prevents the ingress of particles and operates to protect the product. It is a requirement that video recordings of air flow patterns are produced, and these should be considered when establishing the environmental monitoring program.
Clean Air and Clean Air Equipment Qualification
The title of this section has been revised to include the word qualification. So, cleanrooms now need to be qualified and classified and a distinction is made between these terms. Qualification being an encompassing term to describe the full suite of tests to demonstrate the effective control of the environment including viable and non-viable particle monitoring filter integrity testing, air flow visualisation as well as physical parameters like temperature and humidity monitoring. Classification being a term applied to the measurement of non-viable particle monitoring. In terms of defining locations reference is made to the ISO14644 series of standards for guidance in conjunction with an assessment of critical locations in the A/B zone. The guideline makes it clear the non-viable particle classification and microbial qualification be performed in both the at rest and in operational state. Limits are provided for microbial monitoring during qualification with the main change here being that for grade A the requirement is no growth on recovery media (air, settle or contact plates) rather than the <1cfu specification previously stated. Minimum test requirements for requalification are provided including airflow volume, non-viable counts, air pressure measurements and integrity testing of filters as the tests defined in 14664-2 are informative. Scheduled test frequencies for requalification are defined for Grade A/ B areas and grade C/D areas and examples are provided for when non-scheduled requalification would be warranted, for example after a major change within the cleanroom. The requirements for cleanroom qualification and classification are reviewed in detail in our Cleanroom Principles in Practice Course which includes demonstrations of the test instruments utilised during these activities.
Transfer of Materials and Personnel
There is more emphasis regarding understanding and controlling the potential routes of contamination through the facility with enhanced requirements for material and personnel transfer. These include greater detail around the need for segregation of personnel and materials, the transfer of personnel through D to B areas and the need for hand washing in the first stage change room. Increased control around the transfer of materials into Grade A/ B Area with the new requirement that the materials need to be on an approved list and transfer hatches to be protected by effective flushing with filtered air. Where possible materials entering the aseptic areas should be sterilised using autoclaves or depyrogenation tunnels or introduced using an effective transfer disinfection system and gases or liquids must pass through a bacteria retentive filter.
Disinfection
The title of this section has changed from sanitation to disinfection along with current industry terminology. There is a new requirement here for all disinfection processes to be validated with the studies demonstrating the suitability and effectiveness of disinfectants in the manner they are used. The requirement for the use of more than one type of disinfectant agent remains with clarification of the use of a periodic use of a sporicidal agent. Long standing requirements for detergents and disinfectants to be sterile if used in an A/B area remain.
Equipment
This Section has been revised to clarify the need to have a detailed description of equipment design during validation and to maintain an up to date record of P&ID’s. Essentially companies need to ensure that all changes are captured during the lifecycle of the equipment. In terms of monitoring equipment performance, the methods should be defined during the URS stage, with alarm and system trends being reviewed during operation. Cleaning processes should be validated. A detailed assessment of cleaning validation and cross contamination including the application of risk management can be found in our Current Requirements for Cleaning Validation Course
Utilities
In the 2008 Annex 1 guide there were only two clauses related to Utilities. The latest revision has been considerably enhanced and includes specific requirements for the following
- Water Systems
- Steam
- Gases and Vacuum systems
- Heating and Cooling systems
In general, a risk assessment is required to identify high risk utilities (product contact) with greater emphasis on monitoring and maintaining theses. The text on water systems in the second draft has been revised since the first draft with overall requirement that the water produced must comply with the current monograph of the relevant pharmacopeia.

There are numerous references to mitigate biofilm formation within the system for example maintenance of turbulent flow. In line with the current European pharmacopeia WFI may be produced from non-distillation methods.
The requirement for sterilisation of vent filters on water tanks made in the first revision remains as well as a new requirement for the periodic sterilisation or disinfection of the water system. If the word sterilisation remains in the text it becomes problematic as it would be impossible to demonstrate this without the use of biological indicators and the need for this is questionable. There are new requirements for microbial monitoring of the system including the need to determine worst case locations and the need to sample the point at the end of the distribution loop each day the water is used. Finally, for WFI systems the new requirement is that water systems should include continuous monitoring systems like TOC and conductivity unless justified. At Honeyman Training our Pharmaceutical Water Systems Principles in Practice Course has been updated to include detailed discussion of the changes and the application of online monitoring including rapid methods
For steam used as a direct sterilising agent the quality of the feedwater to the steam generator has been clarified as appropriately purified and the steam condensate must meet the current monograph of the pharmacopeia for WFI. The physical parameters for steam quality testing (non-condensable gas, superheat and dryness fraction) shall be periodically assessed. Gases used in aseptic processes must be filtered with a sterilising grade filter at the point of use.
Personnel
This section of the guideline has been revised and expanded to include the requirement for sufficient suitably qualified, trained personnel with experience of the manufacturing technologies to ensure compliance with GMP for sterile products. There are additional requirements around gowning and monitoring including annual initial qualification of personnel along with gowning assessment and specific requirements regarding training of cleanroom personnel depending on criticality of the work performed.

The guideline defines specific elements of the training to include
- Basic Microbiology
- Hygiene
- Cleanroom practices
- Contamination control techniques
- Protection of sterile products
- Airflow visualisation studies
There are enhanced requirements around the garment lifecycle including regular checks for garment quality and integrity as well as the requirement for the use of sterile eye coverings. All these elements related to personnel are included in detail in our Aseptic Processing Principles in Practice Course
Production and Specific Technologies
The revised text includes a small section on Terminals sterilisation, which is virtually unchanged from the 2008 text. The section concerned with aseptic processing has been considerably enhanced.
Aseptic Processing & Preparation
The guideline requires that all aspects of the aseptic process are understood and considered in the CCS to effectively minimise contamination risks. This is the focus of our Risk Management of Microbial Contamination in Cleanroom Operation Course where the risk of contamination to the product is quantitatively assessed through the control measures in place.
Consideration should be given to the use of technical measures to protect the product like the use of barrier technology and engineering solutions to reduce critical interventions and maintenance of grade A conditions during the transfer of materials/components. There are also new requirements for determining validated hold times for sterilised equipment, filtration of product, filling time and maximum time open containers are exposed in the critical processing zone.
Finishing of Sterile Products
There are enhanced requirements around sealing and capping including maintenance of environmental conditions and closure integrity validation which should consider transport and shipping. For inspection of the product there are new requirements for defect classification, the need to maintain a defect library which captures all known classes of defects and the need for trending results.
Sterilisation
Some long-standing requirements for sterilisation processes have remained, for example the requirement for annual revalidation of heat sterilisation processes. There are additional requirements around selecting appropriate biological indicators for the qualification of sterilisation processes as well as enhanced requirements surrounding the preparation and packaging of materials for sterilisation. For the sterilisation of reusable components manufacturers need to consider the maintenance of grade A conditions during sterilisation steps through appropriate choice of wrappings and transfer into the grade A zone.
For surface steam sterilisation of components there are new requirements for assessing the effectiveness of the process during validation including calculation of equilibration time and temp/pressure correlation. Also, requirements for verification of system performance like leak testing (normally weekly) when the cycle contains a vacuum phase and a method of ensuring adequate air removal (like an air detector). On completion of the cycle load dryness should be assessed through visual inspection.
The requirements for dry heat have been enhanced around the need to understand critical process parameters during validation. The longstanding requirement to demonstrate the effectiveness of a depyrogenation process during validation through a 3-log reduction of endotoxin indicators remains. Examples of how the sterilisation steps fit into the overall contamination control strategy as well as examples of how to conduct risk assessments and identify process improvement opportunities are included in detail in our Sterilisation Principles in Practice Training Course.
Filtration
For filtration of products which cannot be sterilised there is a need to fully understand the filtration step as this is a critical control point in the aseptic process. The filtration strategy should be included in the CCS along with an assessment of the feasibility of redundant sterile filtration. Consideration should be given to the critical parameters or process conditions during validation of the step including the use of product and the selection of the most appropriate bacterial challenge organism during retention testing.
In terms of integrity testing of the filters the guideline requires verification of the sterilised filter assembly before use and after use during post integrity testing. The text also provides manufacturers an opportunity to use risk assessment to justify methods other than PUPSIT to verify filter integrity due to process constraints.
In this situation manufacturers are asked to consider all the contamination risks throughout the supply chain, the methods of integrity testing and levels of product bioburden.
Form Fill & Seal
The new sections on form fill seal operation require a thorough understanding of critical process parameters associated with seal integrity during validation and that seal strength be monitored routinely.
Lyophilisation

As an increasing number of medicinal products produced are now lyophilised biological products there is a new dedicated section to the topic. The guideline requires that all aspects of the lyophilisation step are considered in the CCS including cleaning, sterilisation, loading unloading vials and capping to ensure hazards are identified with suitable controls in place. Again, similar considerations here as other steps including maintenance of Grade A conditions during transfer of partially closed containers, use of sterilised tools during transfers and potential for disruption to air flow patterns during the use of mobile carts.
Closed Systems
The new section regarding closed systems provides several elements to consider in the CCS, the primary concern being that the system has been suitably designed to ensure integrity. Manufacturers should also ensure the system has been designed to minimise the number of manual interventions and that the system is able to maintain sterility. In terms of defining suitable background conditions to the closed system this again comes down to risk assessment of the integrity of the system and the method of monitoring integrity (e.g. pressure testing or monitoring). The criticality of the process aseptic/ terminals sterilisation should be used to considered when defining and appropriate background area for the closed system.
Single Use Systems
Single use systems are increasingly used in manufacturing especially with for biopharmaceutical processing due to the increased flexibility and reduced cleaning validation concerns. The increased reliance of pre-sterilised single-use set-ups and components places more emphasis on supplier management as the on-site sterility assurance have been transferred back to the supplier. So, companies need to ensure that if the sterilisation steps are being conducted by a 3rd party that the controls and validation would meet the manufacturers own sterilisation policies. This is a key element to consider in the CCS strategy.
Potential hazards to consider here are handling fragile systems, management of leaks and holes, set up complexity, maintenance of integrity and any detrimental impact on product quality from surface to product contact. The use of closed systems and single use systems are covered in detail on our Biotechnology Principles in Practice course.
Viable and Non-viable Environmental and Process Monitoring

Considering that the highest percentage of GMP deficiencies relate to quality systems, documentation and investigations it is not surprising that the requirements around this have been considerably enhanced. The underlying emphasis in this section is for the need for useful monitoring which produces meaningful data which can be trended, reviewed and acted upon to ensure the manufacturing operation is under continuous control. Environmental monitoring can only provide a snapshot of the manufacturing conditions due to the inherent limitation of the monitoring methods, so it is important that the suitable instruments and appropriate media are selected to minimise inaccuracies in results achieved.
It is a requirement that the environmental monitoring plan be determined through risk assessment. Considerations include
- Sampling locations (Qualification and Routine)
- Frequency of monitoring
- Monitoring method (effective and does not introduce contamination)
- Incubation conditions
- Historical data and significant changes
- Air visualisation studies
- Trend analysis, Alert and Action levels
One of the key requirements is to really understand the data and to take appropriate actions in the event of increased numbers of alert/ action level breaches or consecutive breaches or changes in the microbial flora within the facility. Regulators have been very clear that when environmental monitoring data from recall events has been subsequently reviewed after the event there is often a significant shift in the microbial profile within the D and C grade areas, which if detected at the time may have prevented the product contamination event occurring.
Non-Viable Monitoring
The continued need to monitor the 5-micron particle in the Grade A zone carries through. Further clarification is provided around continuous monitoring of the Grade A environment with the requirement to monitor the duration of the critical processing including set up. The guideline provides clarification that the sample volume does not need to be the same as for classification and the sample volume requires justification. Finally, the revised text includes the need to review all the data generated through initial qualification and routine monitoring to verify appropriate alert and action levels when in operation.
Viable Particle Monitoring (Microbial Monitoring)
The level of monitoring unsurprisingly comes down to the risk of contamination at the process step with an increased level of monitoring in Grade A/ B areas in aseptic processes. The objective of the microbial monitoring plan is to determine suitable locations within the manufacturing operations which provide meaningful data to provide assurance that appropriate environmental conditions were maintained. The monitoring locations need to be close to the action so a review is required of the operations within the manufacturing areas to identify high traffic areas, hot spots, potential dead zones, critical contact surfaces, high traffic areas and surfaces within the cleanroom. The CCS should consider all the potential sources of contamination and transfer routes throughout the facility to focus the monitoring locations on the most critical operations. Enhanced requirements around personnel monitoring have been added with consideration to monitoring personnel following critical interventions and on exit from the Grade B area (gloves, gowns and sleeves). For microbial monitoring of air, the efficiencies of the sampling methods require qualification. The section also includes guidance on the implementation of rapid or automated methods after demonstration of equivalency or superiority to the established methodology (traditional culture-based methods).
The action limit for detection of microbes in Grade A zones has been revised to ‘no growth’ and clarification has been provided that the growth of any microbes from monitoring within the grade A zone requires investigation. Clarification is also provided on the need to identify microbes recovered in the Grade A and B areas to species level and that consideration should be given to the identification of microbes recovered in C and D areas in out of trend or Out-of-Specification (OOS) events.
Aseptic Process Simulation (APS or Media Fill)
The 2008 text contained 5 clauses to address the subject of media fills, concise to say the least for such an important aspect of aseptic processing. This section has now been expanded to 19 clauses with requirements on how to conduct the trails, aseptic manipulations, interventions and actions to take in the event of a media trail failure.
In terms of conducting the media fill a plan is required to determine the challenges to be performed as well as the need to simulate the actual process as closely as possible including factors like shift change-overs. A requirement for the minimum number of containers to be assessed through risk assessment to ensure that all activities and interventions are included, typical minimum numbers of 5,000 and 10,000 units (the former for small batches) remain unchanged. Further guidance is provided around incubation of filled units and identification of microbes recovered from contaminated units. It is very clear from the guidance that the target during a media fill is zero growth. In the event of recovery of a contaminated unit an investigation is required, which determines the most probable root cause, the associated corrective measures, the plan for repeating the trail and of course an assessment of all associated records since the last successful media fill. A more in-depth review of aseptic processing and media fills is included in our Aseptic Processing Principles in Practice course.
Quality Control (QC)
This section opens with requirement for the need of appropriately trained staff with experience in microbiology during the design and operation of a sterile manufacturing process. There are many situations where trained microbiologists are required to lead investigations and assess microbiological data, however it is beneficial that all personnel involved with sterile product manufacturing to have basic training in microbiology. This will enable engineers to assess facility/ equipment design or non-microbiologists and actively participate in investigations or have opinions on microbiological data. These aspects are covered in our Microbiology for Non-Microbiologists course aimed at providing all personnel with a sound understanding of pharmaceutical microbiology and the methods utilised during monitoring and testing.
Any assessment is required for all raw materials, components and products where the risk of microbial contamination has been identified in the CCS. There are new requirements for periodic monitoring of bioburden for terminally sterilised processes even if overkill methods have been adopted. For terminally sterilised product a new requirement has been added for sterility test samples to be taken from worst case locations. Finally, written plans are required regarding actions to take when environmental monitoring results are out of trend or OOS.
Summary & Conclusion
For anyone who has been involved in the review of Annex 1 it is evident that this is a rewrite and not just a simple revision. The consultation process is still on going and the final document has not been published, however the consultation drafts provide a very good indication of what the final document will include. To the question are the clauses any clearer to understand or less ambiguous? The answer largely comes down to the interpretation and perspective of the reader. The 2008 text contained 127 Clauses and when you examine these there was often very little detail or clarification to support the requirements. The revised text contains nearly 300 clauses with a much higher level of detail and is written like a road map of the elements to consider in the CCS for sterile product manufacturers and regulators to follow. It is clear from the revised text that regulators are expecting sterile product manufacturers to really understand their process its entirety and to be able to demonstrate adequate contamination control through risk assessment and effective documentation.

EU GMP Annex 1 Manufacture of Sterile Medicinal Products Revision 2020
Download PDF
Expanding from 127 clauses in the 2008 revision to 300 clauses in the 2020 revision, will Annex 1 be any less ambiguous? Clearly the latest consultation draft is more than a revision, it is a rewrite.
The revised guidance requires companies to formally document a contamination control strategy which considers all the technical and organisational measures in place to ensure sterile products meet their critical quality attributes.
This review document is intended to provide a summary of major changes and enhanced requirements in an easy to read format.
About the Author
Marcus Booth is the Training and Consultancy Director at Honeyman Training & Consultancy Ltd and is responsible for managing the development and delivery of Honeyman Training Programs. Marcus has worked in within the field of sterile product manufacturing for 20 years and is the Sterilisation Principles in Practice course leader. Honeyman courses are constantly revised and updated in response to regulatory & standard updates as well as our interaction with our global client base. For further information on any of our training courses please contact us.
About Honeyman Training & Consultancy Limited
With headquarters in the UK, the Honeyman Training is one of Europe’s leading independent experts in- and provider of pharmaceutical and biotech process and support.
With over 30 years’ experience in helping pharmaceutical and biotech companies meet their regulatory obligations and quality needs, Honeyman Training prides itself on being a reliable and pivotal partner to its clients.
Contact us
Get in touch
Testimonials
Training prospectus 2024
We have produced an overview of the course information available online, available here in a handy PDF Prospectus.

Our Customers
Our team has trained and works with the world's leading Pharmaceutical companies, including …














Latest News





1st December 2023
Why Attend the Microbial Risk Management During Cleanroom Operations Course?
Read more


29th September 2023
Benefits of Microbiology Course for Non-Microbiologists in Pharmaceutical Manufacturing
Read more
